

FDA失速?生物药审批“欧洲优先”背后的监管困局
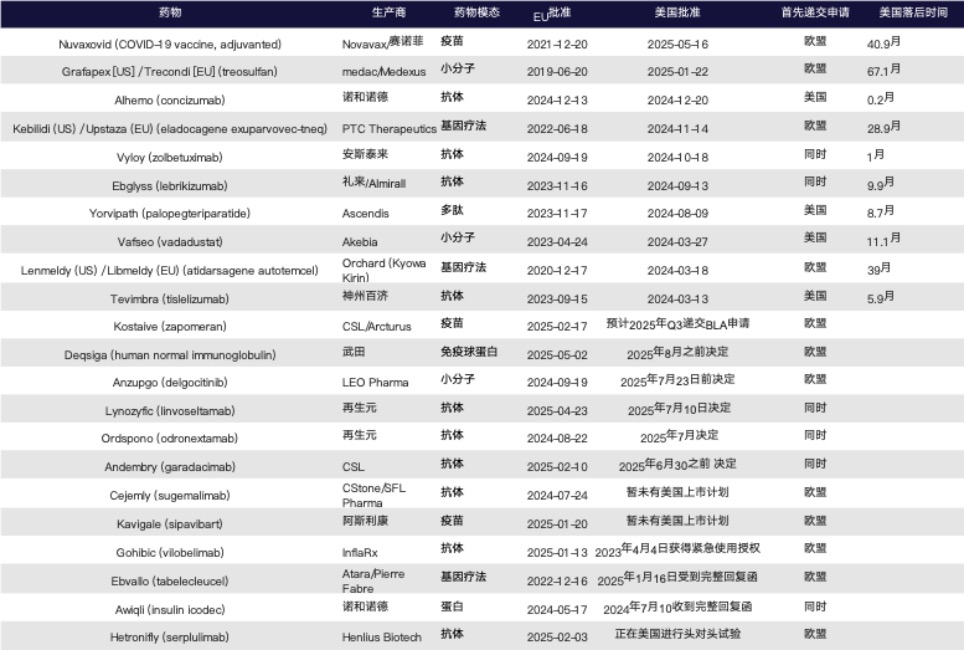
表 1. 2024年至2025年5月20日欧盟领先美国上市新药一览表
以典型案例来看,Novavax的COVID-19疫苗Nuvaxovid早在2021年底就已获得欧盟批准,美国直到2025年5月才完成正式批准,时间滞后超过40个月;medac/Medexus的Grafapex(treosulfan)在欧盟于2019年获批,美国则在2025年初才通过,滞后长达67个月。类似的还有Atara的Ebvallo(tabelecleucel),其在2022年即获欧盟上市许可,至2025年仍未获得FDA批准,反而收到CRL,主要因制造流程未满足FDA对一致性和GMP的高标准要求。
在2025年欧盟先批的新药中,有3款用于新冠防治,包括Arcturus的Kostaive(首个自扩增 mRNA疫苗)、阿斯利康的Kavigale(长效抗体),以及InflaRx的Gohibic(ARDS治疗用抗体)。虽然这些产品在欧盟获得上市,但在美国则未能获得常规BLA批准,甚至部分尚未递交上市申请,反映出制药企业对美国监管节奏及市场预期的重新权衡。
进一步观察数据还发现:除了极端的时间滞后案例,诸如PTC的Kebilidi(延迟28.9个月)、Orchard的Lenmeldy(延迟39个月)、Akebia的Vafseo(延迟11.1个月)等产品,也呈现出“欧先美后”的审批路径。这种时间差的背后,除了临床数据与适应症设定外,核心在于FDA对制造数据、生产体系一致性和第三方代工厂的深度审查,这些都大幅拉长了美国市场的准入周期。
总体而言,美国FDA在新分子实体方面仍占据审批数量和速度的双重优势。但欧盟在生物制剂审批方面的“抢先”现象愈发频繁,且集中于制造复杂、CMC挑战高的产品类别。这种结构性变化表明,美国的全球首发地位虽未动摇,但已开始面临来自欧盟在特定技术领域上的有效挑战。
据统计,2024–2025年欧盟先批的22款药品中,有8款在FDA因制造相关问题收到完整回应信(CRL),其中5例直接关联第三方生产设施的质量问题。来自礼来(Lilly)的lebrikizumab(Ebglyss)便是典型案例:FDA要求其聘请第三方重新评估生产数据的可靠性,而欧洲则在早于FDA近一年时间内给予上市许可。
又如Atara的复发或难治性EB病毒阳性移植后淋巴增殖性疾病细胞疗法Ebvallo(tabelecleucel)在欧洲通过“特殊情况”通道获批后迅速上市,而FDA因GMP问题和第三方厂商不合规发出CRL,并一度对临床试验实施临床暂停。
此外,FDA对于海外生产数据的采信门槛亦显著提高。例如,PTC的基因疗法Kebilidi因临床研究样品与商业化产品工艺不一致被要求补充新的临床数据,导致美国上市时间晚于欧盟两年有余。
某些药物即便在日本或欧盟等海外市场已有多年上市经验和临床安全记录,仍可能因潜在的器官毒性问题而遭遇FDA的严格质疑。例如,Akebia Therapeutics 开发的口服HIF-PHI药物 Vafseo(vadadustat)早在2020年即在日本获批用于肾性贫血治疗,并在2023年获得欧盟上市许可。然而,FDA在审评过程中高度关注其早期临床试验中出现的肝脏不良反应,认为现有数据尚不足以排除肝毒性风险。在经历一次完整回应信(CRL)后,企业不得不收窄适应症范围,仅限于透析依赖型患者,并配套长期肝功能监测方案,最终才于2024年在美获批。这一过程反映出,即便拥有海外上市经验,美国监管方依旧对潜在毒性保留更高警惕性,特别是在作用机制新颖或靶点复杂的药物中更为常见。
对于依赖复杂检测指标作为关键终点的产品,FDA尤其强调原始数据的可追溯性与逻辑一致性。以treosulfan(商品名为Grafapex)为例,该药在欧盟早于2019年便已获批用于造血干细胞移植前的预处理,但其美国新药申请于2020年提交后,连续两次遭遇FDA完整回应信(CRL)。FDA指出,该药关键终点为“无事件生存期(EFS)”,但企业最初提交的数据包中缺少全部患者的骨髓和血细胞检查原始记录,无法支撑终点判定。这一问题使得监管机构在2021年CRL中明确表示“无法确认EFS结果的可信度”,并在随后的补充回复中继续批评数据缺失与逻辑混乱。最终,企业在2024年提交了系统化的溯源审计材料,覆盖所有受试者的骨髓报告与原始数据后才获得FDA认可。
这一审批过程充分说明,FDA在面对此类数据驱动型药物时,其标准不仅仅是“有效”或“安全”,而是“数据是否完整”、“分析是否透明”、“原始记录是否经得起溯源”。这些标准常常远超其他地区的监管机构,从而导致明显的审批周期延长。这也凸显出FDA在面对临床关键指标时,极度重视数据基础是否扎实、可重复。
这项变革虽有助于提升加速批准的可信度,却也在客观上延缓了部分创新疗法的上市节奏,尤其影响了那些尚未启动确证性试验或临床路径复杂的产品。
再生元开发的CD20×CD3双特异性抗体odronextamab(商品名:Ordspono)便面临这一制度掣肘。该药在欧盟已于2024年8月获得批准,用于治疗两种B细胞非霍奇金淋巴瘤:复发或难治性的滤泡性淋巴瘤(FL)和弥漫性大B细胞淋巴瘤(DLBCL)。而在美国,尽管再生元也同步递交了BLA申请,但FDA于2024年3月向其发出完整回应信(CRL),指出公司所提交的III期OLYMPIA临床试验尚处于剂量探索阶段,未能满足FDORA法规下“确证性试验正在进行”的要求。因此,再生元不得不分拆提交,将BLA缩减为只覆盖FL适应症,而DLBCL部分则延后处理。
这类制度性的门槛,不仅延迟了多发适应症同步推进的可能,也对企业的申报策略、临床节奏与商业部署带来更高的不确定性。
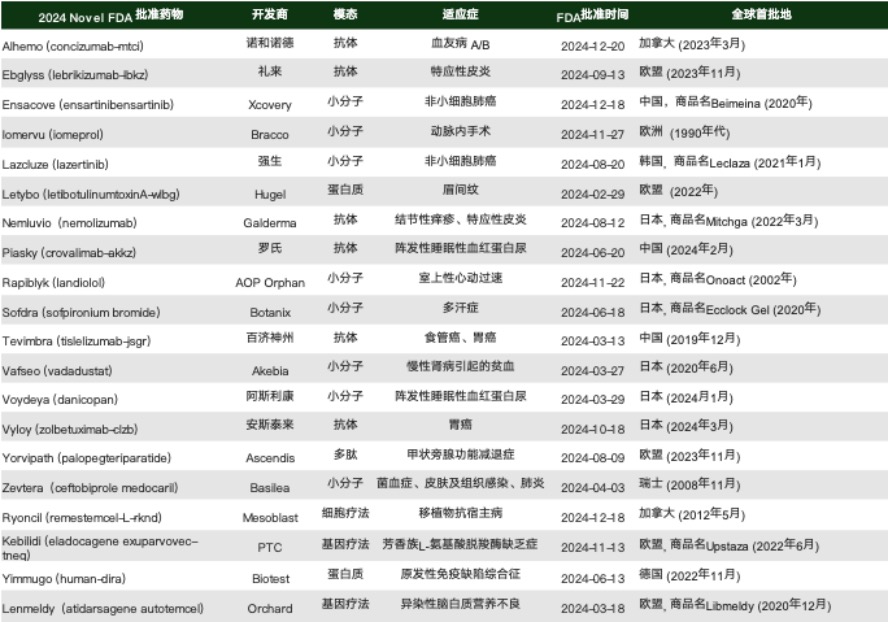
表 2. 非美国首次上市的FDA 2024年批准新药
这不仅反映出这些国家监管机构能力的提升,也显示出全球药企对不同市场审批效率和路径确定性的重新评估与布局。对FDA而言,这种趋势可能进一步加剧其在生物制剂领域的“时间劣势”感知。
从全球新药审批格局来看,“美国首批”已不再是唯一常态,首发地的多极化趋势正在成为行业新现实。
这并非意味着FDA正在“放慢脚步”,而是反映出其对制造一致性、毒性控制、数据可追溯性和制度规范的多重叠加标准,正成为全球最严的准入门槛之一。随着FDORA等法规的强化实施,“快速上市”的定义在美国变得更为谨慎和多条件绑定。
与此同时,EMA的策略则体现出更大程度的灵活性:在制造问题尚可控、数据充分支持疗效的基础上,以有条件批准、例外路径等方式为高需求治疗选项开通通道。这种对风险与应急之间的动态平衡,正成为欧盟在新药首发竞争中频频“超车”的关键变量。
对全球药企而言,未来的新药首发路径不再只是技术成熟度与市场潜力的博弈,更是对不同监管体系中“可控性”与“可预测性”的评估。在这一新格局下,制造体系的稳健程度、注册策略的全球统筹能力,以及企业自身的临床开发节奏掌控,将共同决定创新药物能否在全球多个市场高效落地。
首发之争的焦点,已从审批速度的简单比较,转向了对监管适应能力的全链条挑战。

文章评论(0)