

礼来新冠中和抗体获FDA紧急使用授权,2020年底前将生产100万剂
辉瑞新冠疫苗“Ⅲ期有效性90%”的重磅消息始出,礼来的新冠中和抗体也紧随着迎来了关键性进展。
当地时间11月9日,礼来研发的新冠中和抗体bamlanivimab(LY-CoV555)获得美国FDA的紧急使用授权(EUA),用于治疗近期确诊的12岁以上成人和儿童轻中度高风险新冠患者,这些患者体重至少达到40公斤,并且新冠病毒感染有发展为严重和/或住院的高风险,其中包括年龄在65岁以上或患有某些慢性病的人。

从紧急使用授权书中的内容来看,Bamlanivimab需在确诊后且症状出现10天以内尽快静脉注射,不能用于住院或需要吸氧治疗的新冠患者,同时此次被批准的剂量为必须在60分钟内通过单次静脉注射700毫克。
值得一提的是,Bamlanivimab是在10月7日向FDA递交的EUA申请,到如今获批不过一个月的时间。
1、2020年底前生产100万剂
Bamlanivimab是礼来与AbCellera公司合作开发的用于治疗和预防COVID-19的IgG1单克隆抗体,可以直接结合SARS-CoV-2的刺突蛋白,阻断病毒黏附和进入人宿主细胞,从而起到中和病毒的作用。
据悉,Bamlanivimab获得紧急使用授权的消息一出,礼来公司的股价盘后涨逾3%,不过在这背后,如何快速将Bamlanivimab应用于患者也成为了市场关注的重点。
据外媒报道,美国政府已经购买了30万剂Bamlanivimab,以免费分配给高危患者使用,礼来公司也会立即开始将Bamlanivimab运送到分销商AmerisourceBergen,由其按照美国政府的分配计划进行分配。
此外礼来表示,到2020年底将生产多达100万剂700毫克Bamlanivimab,并将于明年年初起在全球范围内使用,随着产能提高,从2021年第一季度起供应量将大幅增加。
在此基础上,礼来承诺一旦美国政府需要可在2021年6月30号之前另外提供65万剂。以目前30万剂、总价3.75亿美元的政府采购价来算,那么彼时合同采购金额将有可能再增加8.125亿美元。这也意味着,彼时Bamlanivimab的销售额将有可能达到10亿美元。
2、Bamlanivimab研究回顾
FDA表示,此次bamlanivimab获得EUA是基于一项名为BLAZE-1的随机、双盲、安慰剂对照的2期临床试验中期分析。该试验在465名患有轻度至中度COVID-19症状的非住院成人中开展, 这些患者在首次SARS-CoV-2病毒检测阳性后3天内,其中101例接受了700毫克的剂量,107例接受了2800毫克的剂量,101例接受了7000毫克的剂量,156例接受了安慰剂。
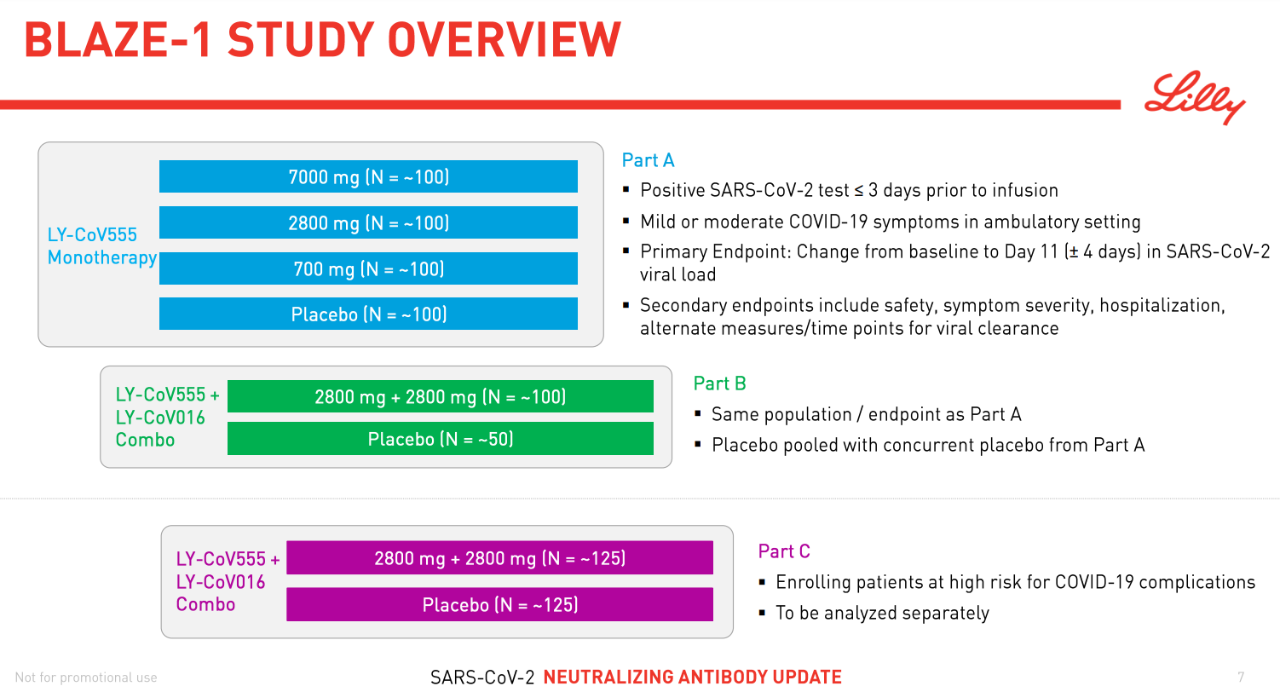
研究结果显示,与安慰剂相比,接受bamlanivimab治疗后28天内COVID-19相关住院或急诊室的就诊人数显著减少。对于疾病进展高风险患者,接受bamlanivimab治疗的患者平均发生住院和急诊就诊的比例为3%,而接受安慰剂治疗的患者为10%。在接受三种bamlanivimab剂量的患者中,对病毒载量以及减少住院和急诊就诊以及安全性的影响相似。
回顾来看,2020年3月12日,礼来与加拿大公司AbCellera就Bamlanivimab研发达成合作,6月2日Bamlanivimab已正式进入临床研究,到8月初,Bamlanivimab已进入三期试验,成为针对新冠肺炎的首个潜在抗体治疗推进了人体试验。
不过围绕Bamlanivimab的临床试验也并非一帆风顺。此前10月份,由于潜在的安全隐患,独立数据安全监察委员会(DSMB)建议暂停礼来Bamlanivimab的另一项ACTIV-3试验,该实验自8月开始进,计划主要在美国招募10000名患者。随后,礼来宣布ACTIV-3临床试验不再招募志愿者,因为根据更新的试验数据,Bamlanivimab不太可能帮助住院的新冠患者从疾病的晚期恢复。
当然,目前Bamlanivimab的其他研究仍在进行中,这些研究专注于早期新冠肺炎患者及其预防,其中包括ACTIV-2临床试验、BLAZE-1和BLAZE-2。
不过值得注意的是,此次FDA授予Bamlanivimab紧急使用权,虽可以在没有足够的经批准的代替药物时,提供帮助诊断、治疗或预防威胁生命的疾病的产品,但该授权终究是临时,不能代替正式的审批流程。这也就是说,Bamlanivimab仍然是研究用药物,尚未获得BLA的批准。
接下来,期待Bamlanivimab的进一步临床数据和好消息。
以上消息综合自第一财经、医麦客、佰傲谷等。
文章评论(0)